






Alzheimer in few numbers
Today, it is estimated that 860 000 people in France and 35 million around the world suffer from dementias of the Alzheimer type. Although its appearance before the age of 65 is rare (0.5%), its frequency is 2 to 4% beyond this age. It then increases proportionally with age, reaching 15% at age 80 years. The disease affects more and more women (1 woman out of 4 and 1 man out of 5, after the age of 85 years). The number of patients in 2020 is estimated to reach two million within the French population alone. Dependency appears 3 to 5 years after the first symptoms appear. It then becomes a medical, scientific, social, but also economic problem, which preoccupies all developed countries in which life expectancy has increased regularly for a century. Alzheimer disease is characterized by a slow neurodegeneration that begins in the hippocampus then spreads to the rest of the brain. This degeneration is the result of the concomitant progression of two types of lesions: on the one hand, an abnormal accumulation outside nerve cells of a protein known as ß-amyloid peptide (also A-beta or Aß peptide) leads to the formation of “amyloid plaques” also called “senile plaques“ ; on the other hand, an abnormal accumulation of the protein TAU inside the neurons causes them to degenerate. The progressive accumulation of these lesions is at the origin of the symptoms of the disease. Memory loss is often the first symptom and orients the diagnosis. The disease then affects executive functions and spatial orientation. Disorders of language (aphasia) writing (dysorthographia), movement (apraxia), behaviour, mood (anxiety, depression, irritability) and sleep with insomnia then develop progressively.
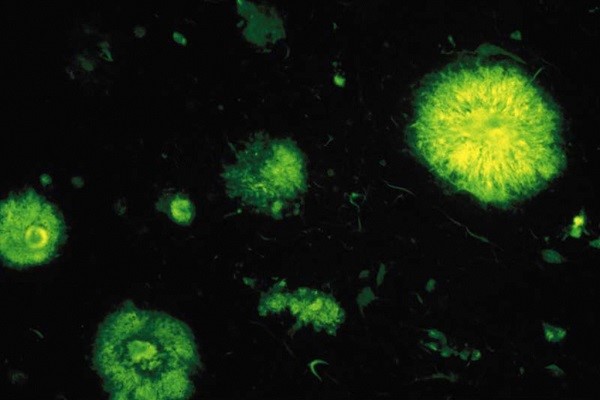
Senile plaques or amyloid plaques observed by fluorescence in a case of Alzheimer disease. They derive from an accumulation of ß-amyloid peptide, consisting of 42 amino acids. © Inserm/U837
To better understand
- The ß-amyloid peptide (A-beta peptide or Aß peptide) is an insoluble protein fragment that cannot be degraded efficiently by the surrounding cells. It accumulates outside the cells, forming “senile” or “amyloid” plaques. The ß-amyloid peptide is neurotoxic.
- TAU proteins are normally present in our organism, in which they facilitate the polymerisation of microtubules (micro-fibrils that constitute the skeleton of cells and guide the transport of molecules in cells and their extensions). TAU proteins play a role in cell regulation and are necessary for stabilizing microtubules. In Alzheimer disease, modified TAU proteins form neurofibrillary tangles, which disorganize the microtubules and cause the death of the neurons.

© JP Parienté for Institut du Cerveau – ICM
Joint studies of Alzheimer disease and prion diseases, why? Alzheimer disease and the prion diseases are neurodegenerative diseases that have in common certain clinico-pathological signs, as well as the mechanisms by which the lesions spread and their neurotoxicity. Both groups of pathologies manifest themselves clinically by dementia, which is most often sporadic, without an apparent cause, but a small percentage has genetic causes. As concerns the lesions, both types of disease are characterized by the presence of amyloid plaques in the central nervous system formed by the accumulation of misfolded proteins. The plaques observed in Alzheimer disease are composed of Aß peptides, whereas those of prion diseases are principally composed of abnormally folded prion proteins called scrapie proteins or PrPsc. ß-amyloid and PrPsc are toxic to the brain and lead to the death of neurons, which is the origin of the principal clinical characteristics of these diseases.
Research at the Institut du Cerveau – ICM
At the Institut du Cerveau – ICM, the team of Stéphane Haïk and Marie-Claude Potier studies prion diseases and Alzheimer disease in parallel, because of the important biological analogies between them. The advances recently made on the prion diseases, the most frequent form of which is Creutzfeldt-Jakob disease, have contributed to a better understanding of the biological mechanisms underlying the development of Alzheimer disease. This research team is particularly interested in the molecular mechanisms implicated in the dissemination of prions: a phenomenon of conversion of the normal protein into a toxic form that induces neuronal death. This phenomenon has also been evidenced in other neurodegenerative disorders like Alzheimer disease; to understand it and try to block it by treatments adapted to these pathologies is a major challenge. To this end, the team has developed cellular systems that facilitate observation of the process and elucidation of the mechanism of neuronal death. They also study the interactions between the prion protein and other major factors implicated in Alzheimer disease, the ß-amyloid peptide and TAU protein, the aggregation of which is responsible for neurodegeneration. By studying how these molecules accumulate outside and inside the neurons, respectively, the researchers try to find a way to reverse the phenomenon. In Alzheimer disease, neurofibrillary TAU lesions are always present, but one can sometimes find them as well in certain prion diseases like the Gerstmann-Straussler-Scheinker disease, a syndrome of genetic origin. Certain mutations in the prion protein cause, indeed a neurofibrillary TAU pathology, which is also the most important lesion in the clinical expression of Alzheimer disease. The team of Stéphane Haïk and Marie- Claude Potier tries to understand how these abnormal proteins induce the neurofibrillary TAU pathology. The propagation of prions is explained by the fact that a misfolded prion protein, rich in ß-sheets, can aggregate, recruit the normal form of the protein, then convert it into the pathological form; the prions can then spread little by little, from neuron to neuron, from one brain region to another, and even from one person to another in the case of contamination or inoculation. In Alzheimer disease, how the Aß and TAU lesions could disseminate in time and space was long misunderstood. Researchers recently demonstrated that prion-type mechanisms also explain the dissemination of ß-amyloid peptide and aggregated TAU, both of which propagate little by little like a prion. A final question that interests researchers is the notion of toxicity. In prion diseases, one knows that toxicity depends on the expression of the prion protein on the surface of the neurons. It has also been shown that the toxicity of an oligomeric form of ß-amyloid peptide (several polypeptide chains) also depends on the presence of the prion protein on the surface of the neurons. The team coordinates the National reference centre for non-conventional transmissible agents, also called prions, and pilots the National network for the surveillance of prion diseases in France. It is noteworthy that very few teams in the country are interested in human prion diseases. As an extension of their studies at the Institut du Cerveau – ICM, the team also performs translational research aimed at exploiting the methods and results obtained by basic research to develop diagnostic and therapeutic approaches thanks to clinical research performed directly with patients. As in many neurodegenerative diseases, a solid research hypothesis implicates a disorder of lipid metabolism. For several years, the team has been interested in cholesterol. Here, it is a question of brain cholesterol, not to be confused with peripheral cholesterol that is part of the composition of cells and implicated in biochemical processes. An increase in the levels of brain cholesterol was observed in patients with Alzheimer disease. This discovery oriented the work of the researchers towards a new direction: in cell cultures developed by the team, they showed that an increase, even transitory, of cholesterol at the surface of the neuronal membrane leads notably to an increase in the production of amyloid peptide. The study of these mechanisms will permit them to go a little further and try to inhibit the effects of these lipids on the production of amyloid peptide. In addition, the team developed, in 2013, a system of cartography allowing for the co-localisation of different molecules in the brains of patients, notably ß-amyloid peptide and brain cholesterol. With this technique, they demonstrated the existence of a high level of cholesterol in senile plaques, probably an indication of a relationship between cholesterol and neurodegeneration. The researchers are now working on mutations in the precursor of amyloid peptides, which will help to better understand the underlying mechanisms. Finally, their studies on brain cholesterol led to the discovery of a modification of a cellular sub-compartment in Alzheimer disease, the so-called endosomal compartment. Endosomes are cell organelles that play a role in sorting and recycling. The endosomal compartment permits fragments of membrane to enter cells; it is at this level that amyloid peptides are produced. The studies conducted on endosomes have incited the teams to do research on trisomy 21. Indeed, people with trisomy 21 have a higher risk of developing Alzheimer disease. At age 60 years, about 45% of those with trisomy 21 have a dementia of the Alzheimer type, whereas in the general population the prevalence at the same age is less than 5%. This research will help understand why these people are more affected by Alzheimer disease.
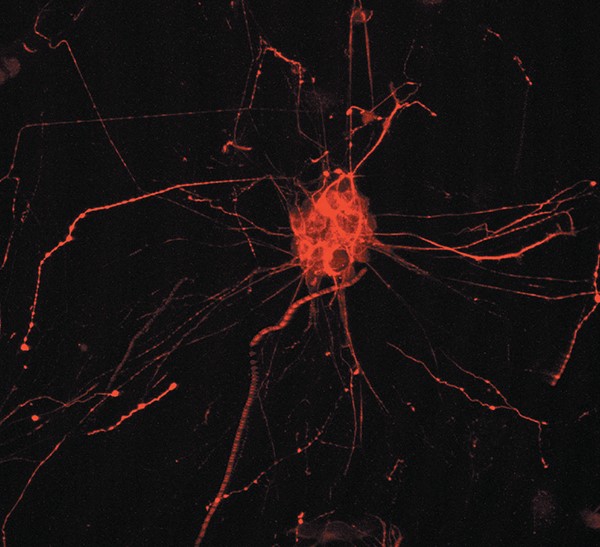
Fluorescent labelling of the protein Tau in a human hNT cell © Inserm/U837
Insight study
INSIGHT, an ambitious world-wide program: The teams of the Institute for Memory and Alzheimer Disease (IM2A) and the Brain and Spine Institute, associated with the IHU-A-Institut du Cerveau – ICM and in collaboration with Pfizer, are carrying out a unique study, world-wide, aimed at better understanding the factors that induce Alzheimer disease: the INSIGHT study. They will follow 400 health volunteers, aged 70 to 85 years, with memory disorders but not Alzheimer disease, with a battery of tests including PET imaging of amyloid. Because of their risk of developing Alzheimer disease, the aim of the study is to observe their evolution and describe the natural history of the disease.

© JP Parienté for Institut du Cerveau – ICM
What hope is offered by research at the Institut du Cerveau – ICM?
At present, the teams that work in the Institut du Cerveau – ICM are particularly motivated to find a way to intervene as early as possible to stop the development of Alzheimer disease. To reduce the time needed for applied research to find a treatment, it is necessary to increase collaborations between clinicians, researchers and patients. It is for this reason that the team of Pr. Bruno Dubois studies subjects at the initial stage of their disease, even the prodromal stage, a period during which a group of precursor symptoms announce the beginning of the disease. In collaboration with the team of Pr. Bruno Dubois, the Clinical Investigation Centre – CIC directed by Dr. Jean-Christophe Corvol performs studies in presymptomatic carriers of genetic forms of the disease; the objective is to be able to make an early, even predictive, diagnosis and develop new strategies targeted to anti-B-amyloid and anti-TAU therapies. Advances in research at the Institut du Cerveau – ICM also concern the identification of new mutations in the gene encoding the prion protein, which cause either classical prion disease or a prion disease associated with neurofibrillary TAU pathology. This new discovery will permit understanding of how amyloid lesions induce the wellknown neurofibrillary tangles responsible for the disease. Recently, the team of Stéphane Haïk and Marie-Claude Potier identified, in collaboration with researchers in other countries, a very particular form of Alzheimer disease characterized by an evolution that is so rapid that the diagnosis of Creutzfeldt- Jakob disease was evoked. They are trying at present to identify the factors that control the rapidity of its evolution. The ambition of the team is to better understand how prions propagate in the nervous system, be they prion proteins or other proteins that aggregate in neurodegenerative diseases, like those implicated in Alzheimer disease. The researchers wish to understand how these misfolded proteins specifically target certain regions of the brain and induce neuronal death. Finally, they use concepts emerging from their studies on prions and techniques developed in their laboratory at the Institut du Cerveau – ICM to better understand how neurofibrillary TAU lesions develop in Alzheimer disease and cause dementia. Finally, they try to apply the technologies they developed for research to applications that are useful for the diagnosis and treatment of brain diseases caused by misfolded proteins in humans.