





The majority of neurodegenerative disorders, from Alzheimer’s disease to amyotrophic lateral sclerosis to Parkinson’s disease, share common features with prion diseases.
These various neurodegenerative diseases are possibly caused by accumulation in the brain of abnormal proteins, misfolded, and toxic to neurons. These pathological proteins propagate step by step from one neuron to another and spread throughout the brain tissue, ultimately driving the death of neurons. This propagation process is called the “prion-like” phenomenon, referring to the mechanisms de-scribed in prion diseases. The “prion model” is thus a model of choice for understanding the biological mechanisms responsible for proteinopathies.
A neurodegenerative disease
Prion diseases, of which Creutzfeld-Jakob is the most common, are rare diseases. They are character-ized by dementia in addition to neurological signs (vision problems, coordination deficits…). Once the first symptoms have occurred, the evolution is rapid and leads to death within a few months. To this date, no standard treatment has been found.
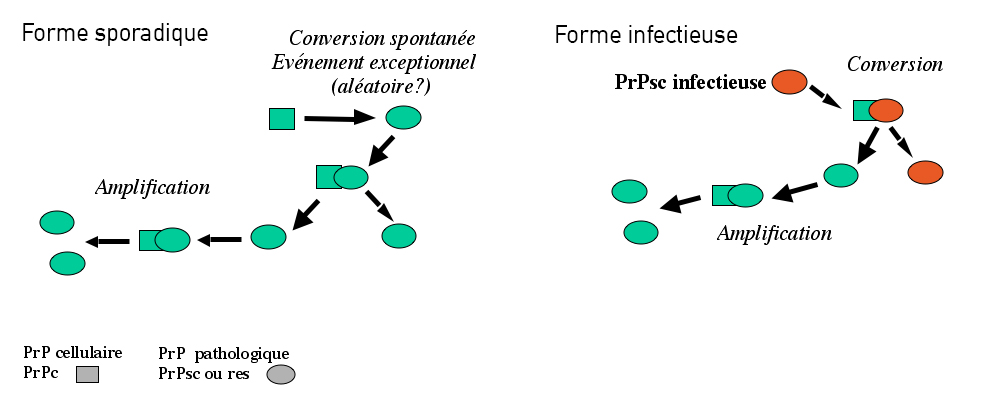
In the majority of cases (85%), the disease occurs randomly, known as a sporadic form. In 15% of cases, the disease can be caused by genetic mutations, and in very rare cases, it can be transmitted during a medical procedure or from exposure of infected tissues by an infected agent (for example, the agent in spongiform bovine encephalopathy that brings about mad cow disease).
An accumulation of abnormal proteins
The prion protein, or PrP is present in most of our cells. While it is strongly conserved across species, its role is still poorly understood. In neurons of diseased patients, prion proteins adopt an incorrect conformation, becoming resistant to degradation and aggregate amongst themselves, becoming “scra-pie” proteins, PrPsc. The deposits of proteins multiply and accumulate both inside and outside neu-rons, leading to their dysfunction, and ultimately, degeneration. The propagation of prions relies on their ability to interact with the normal form of the prion protein and to change the conformation of the normal proteins, converting them into pathological forms.
Prions, Alzheimer’s, Parkinson’s, and more: the same battle
The team of Stéphane Haïk and Marie-Claude Potier studies both Alzheimer’s disease and prion dis-ease because of the biological similarities between the two diseases. The team is interested in, among other aspects, the molecular mechanisms implicated in the propagations of prions, and more precisely, the phenomenon of converting normal PrP proteins into toxic, malformed PrPsc proteins that accumu-late, propagate, and lead to the death of neurons.
Understand and better diagnose
The reseachers developed amplification methods of malformed proteins in vitro in order to observer the misfolding process (incorrect folding of proteins) and to understand its role in the mechanisms of neuronal death. These methods allow, on one hand, studying of the propagation mechanisms and to model the barriers to transmission of these agents, and on the other hand, to develop diagnostic ap-proaches with high sensitivity and specificity and to test therapeutic molecules.
Technology transfer
With this propagation phenomenon having been previously described in other neurodegenerative dis-orders, studies are now in progress to transfer this technology to other proteinopathies (neurodegenera-tive disorders characterized by the abnormal accumulation of certain proteins) of the central nervous system, such as Alzheimer’s disease (accumulation of β-amyloid peptides and of tau proteins) and Parkinson’s disease (agregation of α-synuclein). Understanding this phenomenon is a major challenge for attempting to block it and to find new treatments adapted to these pathologies.
Modeling prion propagation in the laboratory
In order to study the propagation of prions and the consequences of their replication on the survival and the functions of neurons, the team of Stéphane Haïk and Marie-Claude Potier developed different specific models. For example, the first cellular system allowing propagation of different human sources of prions was obtained. This is a precious tool for the research of therapeutic molecules. In-deed, the action of anti-prion treatment often varies in function of origins of the prions, it is thus criti-cal to evaluate efficiency of treatment with respect to prions isolated directly from affected patients.
Identify a new therapeutic target
The cellular models developed by the team were also invaluable in a collaborative study with the group of Odile Kellerman and Benois Schneider at the Paris Descartes University, allowing researchers to propose a new therapeutic target common to prion diseases and Alzheimer’s disease. This work showed the key role of an enzyme PDK1 in the accumulation of pathological proteins, characteristic of prion diseases and Alzheimer’s disease.
In normal conditions, the proteins underlying β-amyloid peptides and the prion protein are cleaved by the enzyme TACE, present at the surface of neurons. The proteins that result from the cleavage by TACE are not toxic for the brain and even play a protective role for neurons.
In neurons infected by pathological prions, as in neurons of patients suffering from Alzheimer’s dis-ease, TACE is no longer localized on the surface of cells, but is sequestered inside neurons. The ab-sence of TACE leads to a faulty physiological cleavage of the non-pathological cellular prion protein (PrPc) or of the amyloid precursor protein (APP) that consequences in production of abnormal toxic proteins.
Another protein, called PDK1, is responsible for sequestering TACE inside neurons. By blocking the activity of PDK1 in experimental models, the researchers were able to block the production of toxic β-amyloid proteins or prions.
PDK1 is thus a promising therapeutic target for future treatments, both for Alzheimer’s disease and for prion diseases.
Establishing clinical trials
The rarity of Creutzfeld-Jakob Disease (1-2 new cases per million of habitatnts), the distribution, and sub-acute evolution of the disease have for a long time limited the ability to perform controlled clini-cal trials, in which in efficiency of a new therapeutic is compared to a placebo.
The largest controlled clinical trial at an international scale (France and Italy) ever undertaken for this disease tested the effectiveness of doxycycline in 2014. Led in France by Institut du Cerveau – ICM researchers (Jean-Philippe Brandel and Stéphane Haïk), the study revealed new perspectives concerning the establish-ment of controlled clinical trials for Creutzfeld-Jakob disease.
In order to be able to propose all suspected patients in France inclusion in a trial, regardless of where the patient was identified, a new recruitment plan and opening of investigation centers has been set up. In two years, this system allowed recruitment of 71 patients in 49 investigation centers of which 27 were opened for emergencies.
Even though no significant effect of treatment was revealed, the study brought major lessons for the evaluation of future treatments that will be proposed for Creutzfeld-Jakob disease:
– Trials controlled against a placebo are feasible for this disease.
– Development of multi-national trials is required in order to be able to recruit a sufficient number of patients in this rare and rapidly fatal disease.
– The original procedure for recruiting patients and opening of investigative centers urgently with ac-celerated administrative procedures established by France could serve as an example.
By relying on this first international experiment and according to the evolution of scientific results, the establishment of a future European trial will be discussed.
Did you know?
Prions are today the only infectious agents devoid of genetic material (DNA or RNA), unlike conven-tional transmissible agents, viruses, bacteria, and parasites.
In 1982, American neurologist Stanley B. Prusiner, put forward the hypothesis that the infectious agent was in fact a protein, and he created the term “prion” (contraction of the term, “proteinaceous infectious particule”). This hypothesis, entirely original and “heretical,” is now widely accepted. Prus-iner was awarded with the Nobel Prize in 1997 for this work.