





La maladie de Huntington est une maladie neurologique génétique qui apparaît généralement à l’âge adulte. Des équipes de chercheurs et de cliniciens au Grenoble Institut des neurosciences (Inserm/ Université Grenoble Alpes) et à l’Institut du cerveau (Inserm/Sorbonne Université/CNRS/AP-HP) ont découvert des anomalies cérébrales dans des cerveaux d’embryons humains porteurs de la mutation responsable de la maladie de Huntington. Ces travaux à paraître dans Science interrogent sur les mécanismes de progression silencieuse de la maladie et sur le moment et la façon de traiter les patients dans le futur.
La maladie de Huntington est une maladie génétique du système nerveux central, rare et héréditaire. Elle se manifeste habituellement entre les âges de 30 et 50 ans par des troubles psychiatriques, cognitifs et moteurs qui s’aggravent progressivement. Elle est due à la mutation du gène codant pour une protéine nommée huntingtine et se transmet sur un mode dit « autosomique dominant » : hériter d’une seule copie pathologique est suffisant pour développer la maladie. En France, environ 18 000 personnes sont concernées : 6 000 présentent déjà des symptômes et près de 12 000 présentent le gène porteur de la mutation mais asymptomatiques.
Les équipes de Sandrine Humbert, directrice de recherche Inserm au Grenoble Institut des Neurosciences(Inserm/Université Grenoble Alpes) et Alexandra Durr, Professeur des Universités – Praticien Hospitalier à Sorbonne Université, à l’hôpital de la Pitié-Salpêtrière, AP-HP et à l’Institut du cerveau (Inserm/Sorbonne Université/CNRS/AP-HP), s’intéressent aux stades précoces de la maladie de Huntington et à la longue période qui précède l’apparition des symptômes. Dans de nouveaux travaux publiés dans Science, elles se sont penchées sur le moment auquel pourraient survenir les anomalies cérébrales.
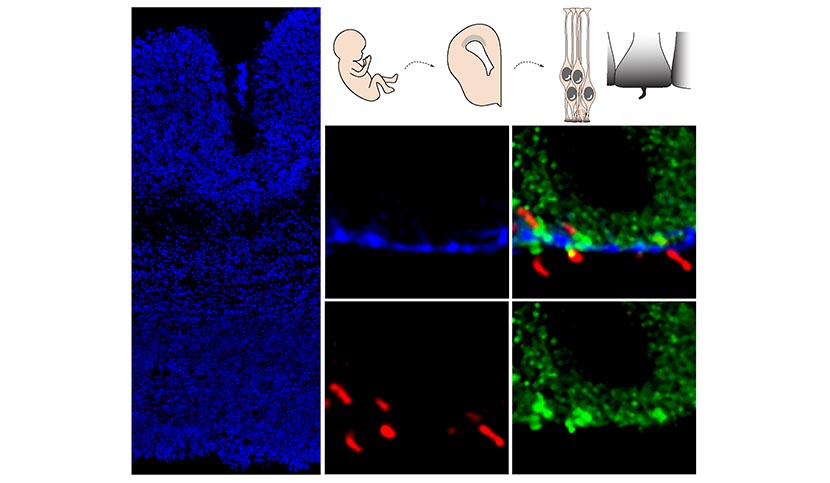
Les équipes de recherche ont étudié des cerveaux d’embryons humains de 13 semaines, issus de dons des parents suite à une interruption médicale de grossesse. Elles ont observé plusieurs différences entre des embryons porteurs de la mutation du gène codant pour la huntingtine et d’autres non porteurs.
Chez les premiers, la protéine huntingtine pathologique est anormalement localisée dans les cellules progénitrices à l’origine des neurones du cortex. Cette localisation anormale est associée, entre autres, à des problèmes de localisation de protéines de jonction dans ces cellules et à des altérations de la taille, densité et orientation du cil, un organelle essentiel au fonctionnement de ces cellules. Ces anomalies perturbent l’équilibre « division-différentiation » des cellules progénitrices. Celles-ci sont en effet issues d’un réservoir de cellules en division dont une partie se différencie en neurones tandis que l’autre continue de se diviser pour fournir de nouvelles cellules progénitrices. Chez les embryons porteurs de la mutation, ces cellules progénitrices entrent plus vite en différenciation au dépend du réservoir de cellules en division.
Les chercheurs ont renouvelé l’expérience avec un modèle de souris de la maladie de Huntington à un stade équivalent de développement embryonnaire et ont retrouvé les mêmes anomalies. Ce travail leur a ainsi permis de valider ce modèle animal pour poursuivre l’exploration des mécanismes précoces de la maladie à d’autres stades du développement embryonnaire ou après la naissance.
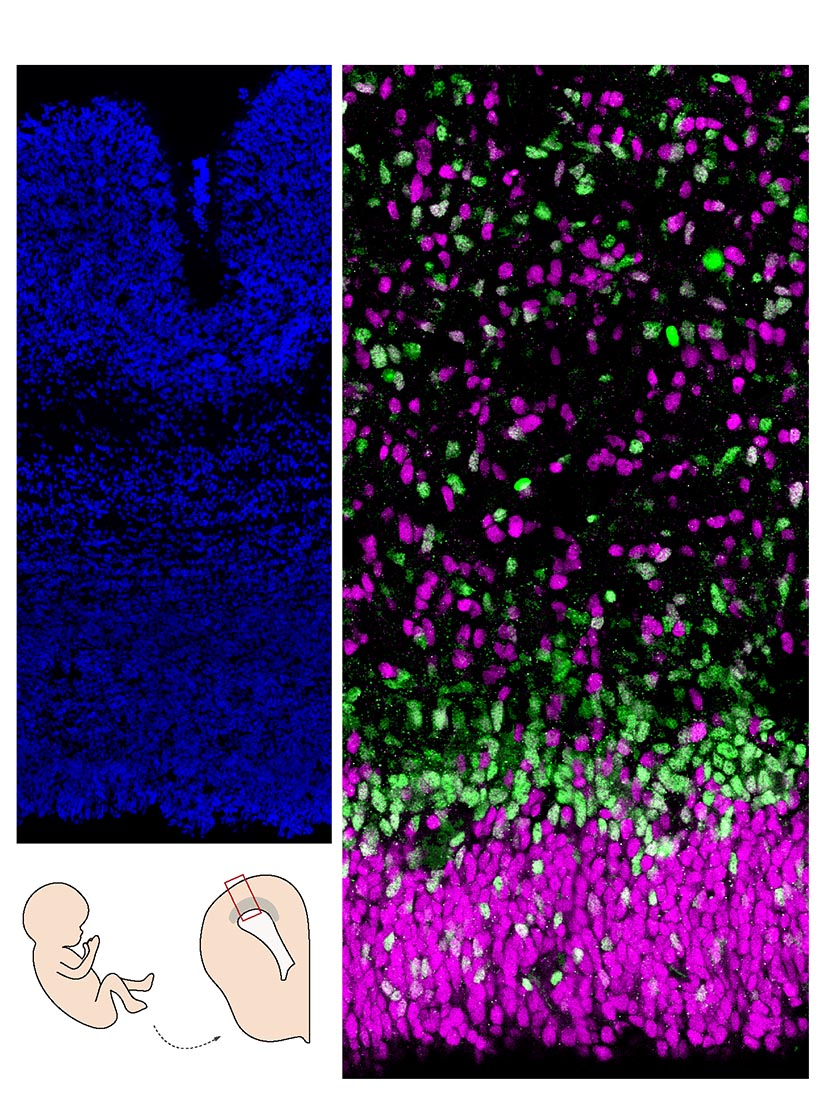
Coupe de cerveau humain (cortex). A gauche, les noyaux sont marqués en bleu; à droite, les cellules progénitrices en magenta sont moins engagées dans la différentiation neuronale que celles en vert. © Monia Barnat/Grenoble Institut des Neurosciences/Inserm, Université Grenoble Alpes
« C’est la première fois que des anomalies du développement cérébral sont mises en évidence dans cette maladie. De plus, celles-ci sont relativement importantes et étendues bien que nous ne soyons pas encore capables de déterminer leurs conséquences directes », clarifient Sandrine Humbert et Alexandra Durr qui ont dirigé ces travaux.
Mais pourquoi les porteurs de la mutation ne manifestent-ils alors aucun symptôme avant un âge avancé ? « À ce stade, nous posons l’hypothèse que le cerveau met très tôt en place des mécanismes de compensation qui permettent un fonctionnement normal. Il se pourrait d’ailleurs qu’il en soit de même chez les personnes porteuses de mutations associées à d’autres types de dégénérescence comme la maladie d’Alzheimer ou la sclérose latérale amyotrophique », précisent les chercheuses.
Celles-ci vont maintenant poursuivre la description du développement cérébral chez des souris modèles de la maladie de Huntington, tenter de comprendre comment ces défauts précoces contribuent à la pathologie adulte, et comment la compensation de ces derniers pourrait être régulée pendant toute la période silencieuse sans symptômes. « Cette découverte a en outre des conséquences importantes sur la façon et le stade auxquels les traitements qui modifient le cours de la maladie doivent désormais être envisagés », concluent-elles.
Sources :
Huntington disease alters human neurodevelopment

